Introduction to Antisense Oligos (ASOs)
Antisense Oligonucleotides (ASOs) are fairly short oligonucleotides, typically 15-30 nucleotides in length, which are composed of either DNA, RNA or a mixture of both. The RNA nucleotides are invariably ribonucleoside analogs that lack a 2’-hydroxyl but still favor a 3’-endo sugar pucker such that the ASO-target duplex adopts a tighter, A-like double-helix structure, which increases the melting temperature (Tm) while also increasing nuclease resistance. The addition of deoxynucleotides in the ASO allows the recruitment of RNase H1 to degrade targeted RNA and may also be used to attenuate duplex stability to reduce off-target effects.
ASOs can leverage diverse molecular mechanisms for modulating gene expression:
- Gene knockdown
- Using RNA-DNA-RNA gapmers that recruit RNase H to cleave the mRNA transcripts
- Using modified RNA ASOs as steric blockers to knockdown translation by preventing the assembly of a functional ribosome on the mRNA transcript by targeting the 5’UTR.
- Gene amplification
- Using modified RNA ASOs as steric blockers to prevent the binding of miRNA to the microRNA Response Elements (MREs) located in the 3’UTR of the transcript. By doing so, the mRNA is protected from the miRNA-induced mRNA decay and the amount of translation of the mRNA is increased.
- Splicing modulation
- Using modified RNA ASOs as splice-switching Oligos (SSOs), both exon skipping and exon inclusion can be induced in pre-mRNA. By targeting the Exonic Splicing Enhancer (ESE), the ASO causes exon skipping by blocking the interaction with stimulatory splicing factors. Conversely, by targeting the Intronic Splicing Silencer (ISS), the ASO causes exon inclusion by blocking the interaction with inhibitory splicing factors.
To successfully modulate gene expression in vivo (and even in vitro for that matter), highly modified ASOs are required which contain modified ribonucleosides and a backbone that is nuclease resistant.
Backbone Modifications
Invariably, ASOs will contain phosphorothioate (PS) linkages in the backbone to improve nuclease resistance. However, PS linkages also dramatically improve cell-uptake, biodistribution and reduce clearance in vivo by increased protein binding. The downside is that ASOs that are highly substituted with PS linkages can cause cyto- and hepatotoxicity. Somewhat counterintuitively, this toxicity is due to protein binding, but not to the plasma proteins, which are crucial to the efficacy of the ASO, but rather to intracellular proteins. In a review by Crooke which focused on PS gapmer ASOs, it was found that toxic ASOs bind paraspeckle proteins and induce the formation of a complex with RNase H1. This leads to mis-localization of the paraspeckle proteins to the nucleolus, inhibition of pre-rRNA transcription and nucleolar toxicity.[1] Flynn et al., expanded upon this work and showed that even fully-modified 2’-O-methyl PS RNAs, which do not recruit RNase H1, still induced nuclear inclusions that contained paraspeckle proteins NONO and SFPQ, leading to increased cell death.[2]
For this reason, there has been considerable effort in finding alternative, non-toxic backbones that can be substituted for the standard PS linkage. Shown in Fig. 1 are two of the most promising alternative backbones to PS – the phosphoryl guanidine (PG), also referred to as PN, and mesyl phosphoramidate (MsPA) and the structures of the PO and PS backbones for comparison. Both MsPA and PG can dramatically lessen PS-induced toxicity while maintaining potency, increasing the therapeutic window.
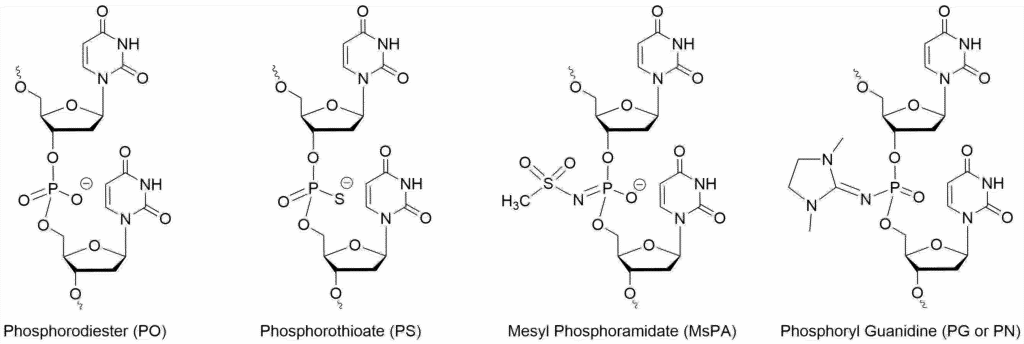
The PG linkage is charge-neutral and, similar to a PMO linkage, extremely stable. In Kupryushkin’s paper which describes the properties of the PG linkage in a gapmer targeting MDR1, the serum stability went from >24 hr for the standard PS modified backbone to >21 days and exhibited approximately 5-fold less toxicity than the PS oligo when the PG linkages were placed in the wings of the gapmer.[3] These results were obtained for an oligo containing stereo-random incorporations of both the PS and PG linkages. However, it’s possible to make a stereopure backbone which can have a profound effect upon the potency of the ASO. In a recent paper, the stereopure PG gapmer was ten-fold more potent than the corresponding stereopure PS ASO.[4] Being similar to the PMO backbone, a fully-modified PG ASO has also been shown to be highly effective as a steric blocker for knocking down translation of target proteins.[5]
The MsPA linkage is negatively charged and, while not as stable as the PG linkage to nucleases, it’s considerably more stable than the standard phosphorothioate linkage. It too can substantially reduce toxicity when substituting PS linkages in ASOs and those substitutions have little effect upon the melting temperature of the duplex. However, rather remarkably, the MsPA is tolerated by RNase H in the gap region, as shown by Anderson et al., though it was found that the number of substitutions should be limited to maintain efficacy.[6] Overall, the MsPA linkage improved the potency of the gapmer while reducing toxicity and increasing the duration of the effect.
Modified Ribonucleosides
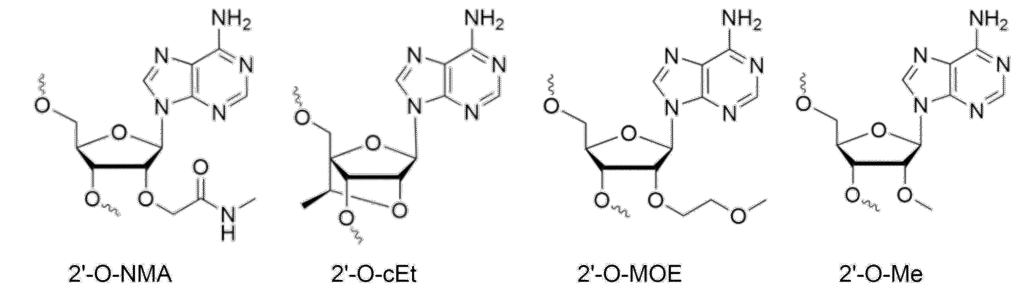
Nucleic acid chemists are an industrious bunch, leading to an enormous number of ribonucleoside analogs whose properties are tailored to optimize duplex stability, minimize toxicity, and increase the potency of ASOs. A recent paper in Science that came out of Robert Britton’s lab described the synthesis of a library of 71 different, diverse nucleoside analogs in a single paper.[7] The vast majority of these modifications are on the sugar rather than the bases. However, there is one very common base modification: 5‑methylcytosine. 5-methycytosine is often used in ASOs for two reasons: 1) the methylation increases duplex stability, and more importantly, 2) it attenuates the innate immune responses of TLR7, TLR8 and TLR9. In this review, we will focus on four of the most widely used analogs in the therapeutic space: 2’-O-Me, 2’-O-NMA, 2’-O-MOE and (S)-cEt, the structures of which are shown in Fig. 2.
The most important properties of modified ribonucleosides are:
- How they affect the stability of duplex
- How resistant they are to exonucleases which are ubiquitous in the cell and plasma
- How toxic they are to the organs in the body
- blood platelets (thrombocytopenia)
- liver (hepatotoxicity)
- kidneys (nephrotoxicity)
- brain (neurotoxicity)
- How they affect the potency of the ASO
Balancing these properties is a subtle art. Take, for example, the constrained ethyl modification, cEt. It is locked into a C3’ endo sugar pucker which is amazingly stabilizing for ASO-RNA duplexes. ASOs modified with these locked nucleic acids will form very stable duplexes, which frequently translates to very high potency. The downside, however, is that this same increase in affinity is frequently accompanied by greater toxicity. For this reason, cEt is used sparingly in ASOs, typically in the wings of gapmers or toward the ends of SSOs.
Previously, this toxicity was attributed to off-target mRNA degradation by RNase H due to the higher melting temperature (Tm) of the cEt-modified ASOs, which made the ASOs tolerant of target mismatches, and therefore prone to off-target effects. However, in an arguably seminal paper by Wen Shen at al., she convincingly argues that the predominant basis for toxicity, is, in fact, binding to paraspeckle proteins such as NONO, SFPQ, PSPC1 and FUS and not off-target effects.[8] Toxic LNA, cEt, and 2’-MOE ASOs cause the paraspeckle proteins to delocalize from the paraspeckle foci to the nucleoli. This causes the paraspeckle foci to essentially dissolve and the nucleoli to undergo stress that leads to activation of p53 and caspases that lead to apoptosis. Interestingly, the delocalization is dependent upon RNase H1 which appears to bind to the ASO-protein complex and shuttle it into the nucleolus. Non-toxic LNA, cEt and MOE ASO bind to proteins with a much lower avidity and do not cause this delocalization. Interestingly, 2’-Fluoro gapmers bind to NONO and PSF much more strongly than MOE gapmers, but rather than causing delocalization to the nucleoli, the proteins are translocated to the cytoplasm where they are quickly degraded by proteosomes.
Therefore, lowering the affinity of ASOs to either RNase H1 or paraspeckle proteins will lead to lower toxicity. As expected, if the ASO is fully substituted with 2’-OMe (gapless) the toxicity is greatly reduced. However, Shen found that gapmer activity could be maintained while lowering toxicity by placing the LNA or cEt in the 3’ wings of the gapmer, due to the apparent preference of proteins to recognize the 5’ terminus of the ASO. However, the most efficacious modification was placement of a single 2’-OMe in the 2nd position of the gap. This lowered toxicity across a broad range of gapmer ASOs while causing a minimal impairment of activity. A group of 328 ASOs, 47% of which were toxic (as measured by a 2-fold increase in caspase activity), were compared to the parent sequences after a single 2’-OMe was placed in the 2nd position of the gap. Afterward only 19% were still toxic afterward.
Shown in Table 1 is our best attempt to provide a summary of the relative values of duplex stabilization, nuclease resistance, toxicity and potency. These values are all sequence- and application dependent – at least to some degree. For example, the NMA is 3 to 4 times more potent than MOE for splice-switching,[12] but in ASO gapmers, they are essentially equivalent. Interestingly, it does not appear that the increased potency splice-switching of the NMA-modified ASO is due to a significant increase in duplex stability or higher accumulation in the tissue due to nuclease resistance; rather it appears the NMA modification may improve the release from the endosome or trafficking to the nucleus.[12]

ASO Gapmers
Gapmers are typically 16 to 26 nucleotides in length, with an internal region of DNA that is typically 10 bases – long enough to easily accommodate the RNase H1 as shown in Fig. 3(A). The ‘wings’ of the gapmer are modified RNA nucleotides which both increase nuclease resistance as well as increase the Tm of the duplex, leading to higher potency. Typically, the canonical design is a 5-10-5 gapmer with 5 bases in the wings and a 10 base internal DNA gap. The Tm can be increased by using two or more incorporations of cEt, allowing the gapmer to be shortened to a 3-10-3 configuration. If the Tm is too high, though, specificity may be lost due to off-target knockdowns. Shown in Fig. 3(B) is a graph depicting the balance of Tm, potency and specificity. If the ASO-target affinity is too high, the specificity will be decreased. Based upon a paper by Papargyri, the optimum Tm to aim for is approximately 62 – 68 °C.[15] As noted earlier, the backbone is typically fully-phosphorothioate modified (PS) which can lead to toxicity, however the addition of single 2’-OMe at the 2nd base in the DNA gap (counting 5’ to 3’) frequently reduces toxicity. Finally, the question remains where to target. If one peruses ASOs with FDA approval or currently in clinical trials, a frequent choice is the 3’UTR. This region remains accessible without ribosomal or spliceosomal machinery blocking access.
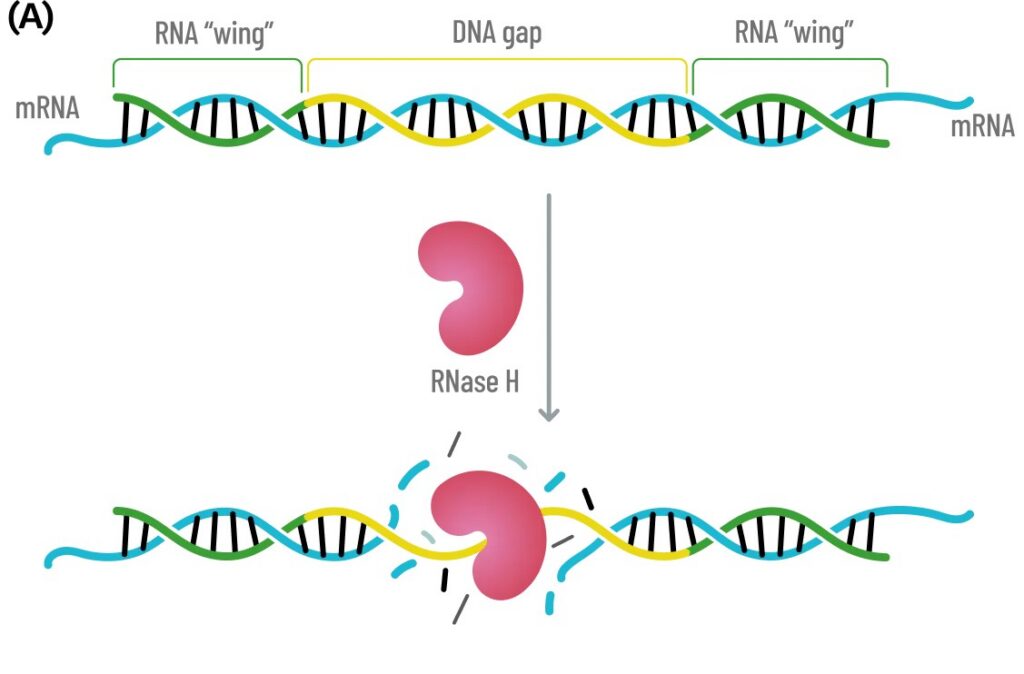
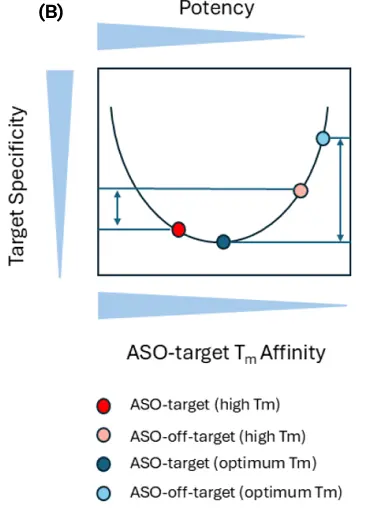
Fig. 3 (A) Schematic of Gapmer ASO hybridizing to target mRNA with the internal DNA sequence shown in yellow and the RNA ‘wings’ shown in green. When the ASO anneals to the mRNA target, the internal DNA-RNA hybrid recruits RNase H, which processively cuts the mRNA transcript. The RNA ‘wings’ increase the ASO stability to nucleases as well as increase duplex stability to the target. (B) Schematic showing the balance between Target-ASO affinity, potency and specificity. If the Tm of the ASO-Target is too high, the ASO will have low specify though high potency. At the optimum Tm, the potency will be high with minimum off-target activity.
Gene Amplification and Knockdown Using ASO Steric Blockers
Steric blocking ASOs are typically uniformly modified with ribonucleotide analogs, such as 2’-MOE, and use a PS backbone to increase stability and uptake. They can both knockdown gene expression as well as amplify it, depending upon what region in the mRNA is targeted (Fig. 4).
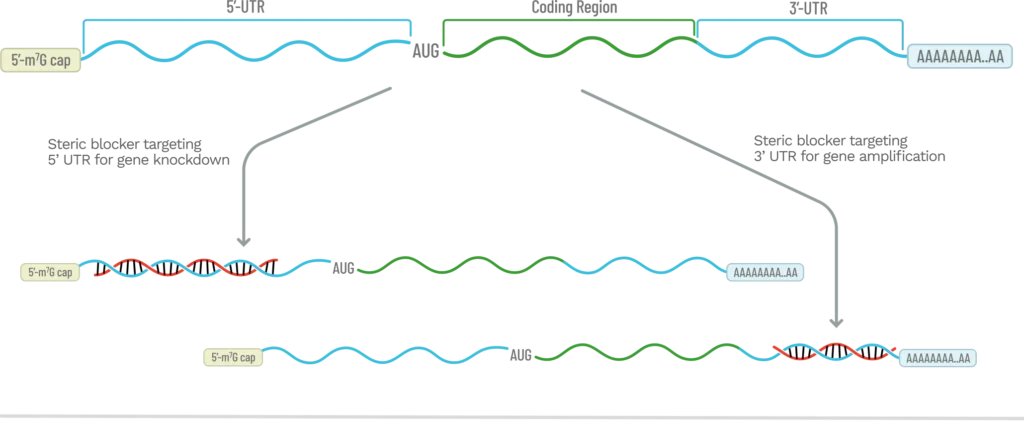
While ASO gapmers are the most widely used method to knockdown gene expression, there are conditions where using an ASO that sterically blocks translation of an mRNA transcript is preferred. RNase H1 only needs a short, six-base DNA duplex to cleave RNA, so mismatches and bulges can be tolerated, leading to off-target transcript degradation and toxicity. Steric blockers knockdown gene expression by preventing the 43S pre-initiation complex in the 5’UTR from binding and scanning to find the AUG start codon. This prevents translation of the mRNA transcript without degrading it. In addition, with steric blockers, mismatches lead to low duplex stability and therefore, the ASO is easily displaced, causing little, if any, off-target knockdown. Finally, because there is no PS DNA gap, the toxicity of ASO steric blockers is greatly reduced.
One of the most effective locations to target using steric blocking ASOs, is just upstream of the AUG start codon in the 5’UTR (or the start codon itself). This will typically cause effective knockdown, however an ‘ASO walk’ will often yield an ASO with higher potency.[16] For therapeutic applications, researchers will often perform a walk with ASOs to find the most potent sequence and then switch over to PMOs since they are considered ‘privileged’ by the FDA given their history of low toxicity.
In the 3’UTR, there are regulatory sequences known as microRNA responsive elements (MREs). These are short sequences that miRNAs bind to modulate translation. While they can be in the 5’UTR,[17] typically they are found in the 3’UTR where ribosomes cannot block access to them. These miRNA-3’UTR interactions almost always suppress gene expression and the commonly cited percentage of protein-coding genes that are regulated by miRNA is 60%.[18] By using a steric blocker to bind to these 3’UTR MREs, gene expression can be substantially increased, with a recent example published by Aggrawal where the miR-29b binding site was targeted with an 18mer uniformly modified with 2’-MOE and a PS backbone linkages.[19] To assist in identifying these MREs in the 3’UTR, there are online tools, such as TargetScan.[20]
Splice-Switching Modulation
Steric blocking ASOs can also be used for splicing modulation of pre-mRNA by targeting either exonic splicing enhancers (ESEs) or intronic splicing silencers (ISSs) to either skip or include targeted exons respectively (Fig. 5).
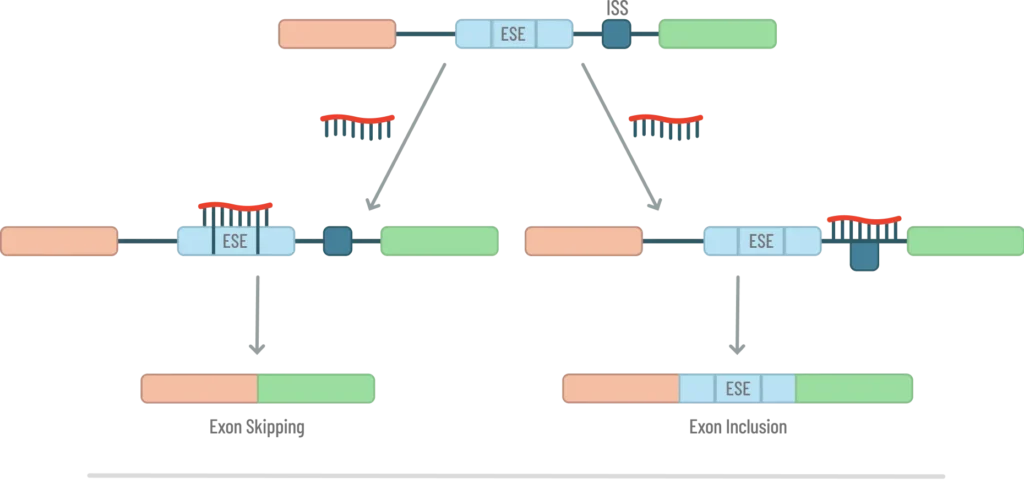
They do this by blocking the interactions of splicing factors that are either stimulatory or inhibitory. In this function, the steric blocking ASOs are called splice-switching oligos (SSOs). While any of the 2’-ribonucleotide analogs will function in the SSO, the 2’-NMA appears to be the modification of choice. A recent paper from Ionis Pharmaceuticals showed that the 2’-NMA modified SSO was 3-4 fold more potent than the traditional 2’-MOE modification despite both SSOs having similar Tm values and concentrations in the central nervous system tissues.[12] Since splicing occurs in the nucleus, one possibility mentioned by the authors to explain the higher potency was that the 2’-NMA modified SSO was trafficked to the nucleus more efficiently. Regardless, this result suggests that 2’-NMA is the modification of choice for SSOs.
ESEs are short sequences found in exons and are typically 6 to 10 nucleotides in length. SR proteins (Serine/Arginine-rich proteins) bind these splicing enhancer elements and promote spliceosome assembly through the recruitment of U1 snRNP and U2AF. [21] There are twelve SR proteins involved in pre-mRNA splicing for both constitutive exons (those exons which are present in all isoforms of a protein) and alternative exons (those which may, or may not be, spliced into the mature mRNA depending upon the cell state). They act as enhancers and targeting these sequences with an SSO leads to exon skipping. ISS elements are 4-18 nucleotides in length and recruit heterogeneous nuclear ribonucleoproteins (hnRNPs) which silence splicing. By targeting ISS sequences, the exon is included in the final mRNA transcript. A good review of the current field of splice-switching therapies including exon skipping, inclusion, pseudoexon skipping and suppression of toxic de novo splice sites caused by mutations, was recently published by Takeshima.[22] To help find ISSs and ESEs to target with an SSO, the Human Splicing Finder is a good online tool for doing so.[23]
You can conveniently order custom ASO libraries through our online store, designed for flexibility and rapid turnaround. To streamline your workflow, Synoligo also offers integrated screening services to help identify high-performing lead candidates. First-time customers can take advantage of our bundling discount for combined library synthesis and screening. Request a quote today!
References
[1] Stanley T Crooke, Timothy A Vickers, Xue-hai Liang, Phosphorothioate modified oligonucleotide–protein interactions, Nucleic Acids Research, Volume 48(10), 5235–5253 (2020) https://doi.org/10.1093/nar/gkaa299
[2] Flynn, Loren L., et al. “Single stranded fully modified-phosphorothioate oligonucleotides can induce structured nuclear inclusions, alter nuclear protein localization and disturb the transcriptome in vitro.” Frontiers in Genetics 13 (2022): 791416
[3] Kupryushkin, M.S. et al., Antisense oligonucleotide gapmers containing phosphoryl guanidine groups reverse MDR1-mediated multiple drug resistance of tumor cells. Mol. Ther. Nucleic Acids, 27, 211-226 (2022). https://doi.org/10.1016/j.omtn.2021.11.025.
[4] Kandasamy, P. et a., Impact of guanidine-containing backbone linkages on stereopure antisense oligonucleotides in the CNS, Nucleic Acids Res, 50(10) 5401–5423 (2022). https://doi.org/10.1093/nar/gkac037
[5] Skvortsova YV, Salina EG, Burakova EA, Bychenko OS, Stetsenko DA and Azhikina TL, A New Antisense Phosphoryl Guanidine Oligo-2′-O-Methylribonucleotide Penetrates Into Intracellular Mycobacteria and Suppresses Target Gene Expression. Front. Pharmacol., 10,1049 (2019). doi: 10.3389/fphar.2019.01049
[6] Anderson, B.A. et al., Towards next generation antisense oligonucleotides: mesylphosphoramidate modification improves therapeutic index and duration of effect of gapmer antisense oligonucleotides. Nucleic Acids Res., 49(16):9026–9041 (2021). doi: 10.1093/nar/gkab718
[7] Anketell, M. J., Fung, E., Liu, W., Shinde, M., He, C. Q., Ng, K. W. C., Silverman, S. M., Campeau, L. C., Pantophlet, R., & Britton, R. A unified platform for nucleoside analog synthesis. Science, 39, 6783, (2026) https://www.science.org/doi/10.1126/science.aed6880
[8] Shen, W, et al., Chemical modification of PS-ASO therapeutics reduces cellular protein-binding and improves the therapeutic index, Nat Biotechnology 37(6):640-650 (2019). doi: 10.1038/s41587-019-0106-2
[9] Scharner, J. et a., Hybridization-mediated off-target effects of splice-switching antisense oligonucleotides, Nucleic Acids Res., 48(2) (2020). doi: 10.1093/nar/gkz1132
[10] Prakash. T.P. et al., Comparing In Vitro and In Vivo Activity of 2′-O-[2-(Methylamino)-2-oxoethyl]- and 2′-O-Methoxyethyl-Modified Antisense Oligonucleotides, J. Med. Chem. 51, 2766–2776 (2008)
[11] Wan, W.B. and Seth, P.P. The Medicinal Chemistry of Therapeutic Oligonucleotides J. Med. Chem. 59, 21, 9645–9667 (2016).
[12] Ling, K. et al., Nucleic Acids Research, 2026, 54, gkag484 https://doi.org/10.1093/nar/gkag484
[13] Seth, P.P. et al., Short Antisense Oligonucleotides with Novel 2′-4′ Conformationaly Restricted Nucleoside Analogues Show Improved Potency without Increased Toxicity in Animal J. Med. Chem. 52, 10–13 (2009).
[14] Pallan, P.S. et al., Structure and nuclease resistance of 2′,4′-constrained 2′-O-methoxyethyl (cMOE) and 2′-O-ethyl (cEt) modified DNAs, Chem Commun (Camb) 48(66):8195-7 (2012). doi: 10.1039/c2cc32286b
[15] Papargyri, N. et al., Chemical Diversity of Locked Nucleic Acid-Modified Antisense Oligonucleotides Allows Optimization of Pharmaceutical Properties, Molecular Therapy: Nucleic Acids, 19, 706-717 (2020).
[16] Johansson H.E. et al., Target-specific arrest of mRNA translation by antisense 2′-O-alkyloligoribonucleotides, Nucleic Acids Research, 22 (22) 4591-4598 (1994)
[17] Liang, Xue-hai et al., Antisense oligonucleotides targeting translation inhibitory elements in 5′ UTRs can selectively increase protein levels, Nucleic Acids Res, 45(16):9528–9546 (2017). doi: 10.1093/nar/gkx632
[18] Diener, C., Keller, A. and Meese, E., The miRNA–target interactions: An underestimated intricacy, Nucleic Acids Research , 2024, 52 (4) 1544-1557 (2024).
[19] Aggrawal, G. et al., Antisense oligonucleotides targeting the miR-29b binding site in the GRN mRNA increase progranulin translation, J. Biol. Chem. 299(12) 105475 (2023).
[20] Vikram Agarwal, George W Bell, Jin-Wu Nam, David P Bartel, Predicting effective microRNA target sites in mammalian mRNAs eLife 4:e05005 (2015). https://www.targetscan.org/
[21] Slišković, I et al.; Exploring the multifunctionality of SR proteins. Biochem Soc Trans 50 (1): 187–198 (2022). doi: https://doi.org/10.1042/BST20210325
[22] Yasuhiro Takeshima, Expansion of Splice-Switching Therapy with Antisense Oligonucleotides, Int. J. Mol. Sci. 26, 2270 (2025). https://doi.org/10.3390/ijms26052270
[23] Desmet, F.-O., Hamroun, D., Lalande, M., Collod-Béroud, G., Claustres, M., & Béroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals, Nucleic Acids Research, 37(9), (2009). DOI: 10.1093/nar/gkp215