In May of 2025, a month after the last dose of k-abe was administered, a remarkable paper was published detailing the conception, development and treatment of the infant KJ Muldoon with an in vivo base editor to cure his carbamoyl-phosphate synthetase 1 (CPS1) deficiency.[1] CPS1 deficiency, which affects approximately 1 in 1,300,000 people, is a urea-cycle defect that leads to dangerously elevated levels ammonia in the blood that, if left untreated, leads to CNS damage and ultimately death. In a spectacular collaboration between industry, academia, and the Children’s Hospital of Philadelphia, researchers designed adenine base editors to repair KJ’s specific mutations. After screening the editors for efficacy and specificity, the team selected a final candidate, k-abe (kayjayguran abengcemeran). The FDA authorized it as a single-patient expanded-access Investigational New Drug (IND), and the therapy was administered to the infant within just seven months of his birth. Below is the timeline from diagnosis to dosage for the base editor (Fig.1).
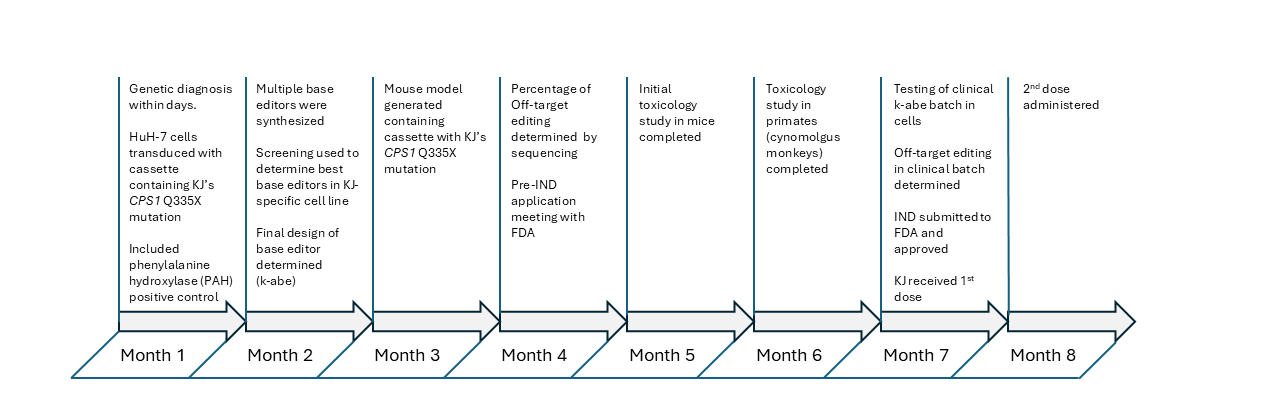
Adenine base editors, like K-abe, were first developed in David Liu’s lab at Harvard.[2] They consist of two components – an mRNA encoding a fusion protein consisting of Cas9 nickase for localizing the adenine base editor to the site of the mutation (in k-abe, CPS1) and the adenine deaminase which catalyzes A-to-I conversion. Due to the nick occurring on the opposite strand, the I.T mismatch is repaired with preferential excision of the T and the insertion of C opposite the inosine. The second component is the guide RNA (gRNA), which directs the Cas9 nickase complex to the specific mutation – in this case, the CPS1 locus for targeted editing. For in vivo editing, the mRNA and gRNA are encapsulated in lipid nanoparticles (LNPs).
The treatment was a success. In a press release given by the Children’s Hospital of Philadelphia at the one-year anniversary of his gene therapy, Baby KJ, while not ‘cured’ per se, was reported to be “thriving”, reaching developmental milestones of children his age, with dietary protein better tolerated and illnesses no longer causing high spikes in ammonia levels.
It appears that the successful treatment of KJ served as the catalyst for the Plausible Mechanism Framework. Just seven months later, in December 2025, FDA Commissioner Martin Makary and the Director of the Center for Biologics Evaluation and Research, Vinay Prasad, published a perspective on NEJM’s Sounding Board that outlined the necessary conditions where the Plausible Mechanism Framework could be applied,[3] with the formal FDA Draft Guidance published two months later.[4] They state that for the Plausible Mechanism Framework to be applied, the following conditions must be met:
- There is a defined, underlying molecular or cellular abnormality that is directly tied to the disease
- The drug or therapy targets the underlying or proximate biological alteration
- There is a known, natural history of the progression of the disease in the untreated population
- There is data that demonstrates the target was successfully drugged, edited or both
- There is an improvement in clinical outcome or course
If these conditions are satisfied, then the Plausible Mechanism Framework may be applied, which greatly reduces the regulatory burden and costs of bringing these bespoke, individualized therapies to the market. This is greatly needed. Between 2018-2024, three different N-of-1 ASOs were approved for compassionate use – Milasen, Valeriasen and Atipeksen – and all were funded by charitable contributions and government grants without any costs being defrayed by insurance companies, giving no incentive for pharmaceutical companies to pursue this class of drugs.
Arguably, the most impactful allowance under the Plausible Mechanism Framework to help ameliorate this issue, is the ability to submit an IND not for a specific molecular therapy, but for a platform. This is possible because of the modular character of these genetic medicines. Using k-abe as an example, it is only the 20 base targeting sequence that homes in on the mutated locus of CPS1. If a different G-to-A mutation leads to the same disease, a different targeting sequence could simply be inserted into the gRNA to edit that locus and make ‘l-abe’ or ‘m-abe’. Under the new guidelines, a separate IND would not be required, and you could cover the range of different mutations under the same ‘umbrella’ master protocol stating:
The Agency is open to the use of master protocols (e.g., umbrella or platform 492 trials) for the evaluation of therapies that target different genetic changes for the same disease [4].
With this new possibility of IND submission under a master protocol, the team that led development of k-abe therapy for KJ is now working on treating seven different urea cycle disorders using prime editing. Unlike a base editors that edits single nucleotides, prime editing can edit a region of gene, editing multiple bases simultaneously.[5] This allows a greater variety of mutations – even distal – to be corrected by either adding or removing a section of the targeted gene. With proof-of-concept data, the researchers requested a pre-IND meeting with the FDA. In their discussions, the FDA agreed with the general design of the master protocol for an open-label, single-arm study spanning all seven urea cycle disorders.[6] This paper was especially interesting in that it reports on their discussions between the researchers and the FDA, and gives additional clarity to the FDA’s approach to implementing the Plausible Mechanism guidelines. For example, they noted that the Chemistry, Manufacturing and Controls (CMC) was quite stringent for an early phase1/2 study typically reserved for late-stage drug development, among other observations.
Under the Plausible Mechanism guidelines, other onerous requirements such as a Random Control Trial, with a large group to demonstrate statistical significance, have been eliminated if the disease – and even the patient, who can serve has a ‘self-control’ – has a documented history of the progression of the disease without treatment. In addition, biomarkers that demonstrate the mutation has successfully been edited, an exon successfully skipped, or the expression of a deleterious protein successfully knocked down, may be used in lieu of clinical results to support the IND. So rather than showing a patient with a long-term disease demonstrably better after the treatment (which often is a lengthy process), biological results can show the underlying cause has been addressed in lieu of a clinical result.
One of main complaints from industry regarding the new guidelines is that new therapies for ASOs, Splice-switch oligomers and siRNAs, are locked into using a limited repertoire of modified bases, so called privileged modifications – e.g. 2’-MOE, 2’-OMe, 2’-F, phosphorothioate (PS) backbone, PMO and a few others – unless large, time-consuming (>1 year) and very expensive toxicological studies are performed. This is unfortunate because phosphoryl guanidine (PG)[7], mesyl phosphoramidate (MsPA)[8], and 2’-4’ constrainted ethyl (cEt)[9] modifications, among others, can improve efficacy, stability and/or reduce toxicity. It is hoped that the FDA will consider lowering the hurdles of introducing new chemistries when the guidelines are finalized. The public comment period will be open until April, 26th 2026 and can be accessed here.[10]
References
[1] K. Musunuru, S.A. Grandinette et al., Patient-Specific In Vivo Gene Editing to Treat a Rare Genetic Disease, N Engl J Med 392:2235-43 (2025).
[2] Gaudelli, N., Komor, A., Rees, H. et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 551, 464–471 (2017). https://doi.org/10.1038/nature24644
[3] Prasad, V. and Makary, M., FDA’s New Plausible Mechanism Pathway, N Engl J Med, 2025; 393:2365-67 DOI: 10.1056/NEJMsb2512695
[4] Plausible Mechanism Framework to Develop Individualized Therapies that Target Specific Genetic Conditions with Known Biological Cause, Draft Guidance for Industry; Docket/Ref: FDA-2026-D-1256; 91 FR 9283
[5] Anzalone, A.V., Randolph, P.B., Davis, J.R. et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157 (2019). https://doi.org/10.1038/s41586-019-1711-4
[6] Feierman, E.R., et al., Implications of the FDA’s new plausible mechanism framework for the development of a personalized in vivo prime editing platform, The American Journal of Human Genetics 113, 1–9, May 7, (2026)
[7] Kupryushkin M, Filatov A, Mironova N , et al., Antisense oligonucleotide gapmers containing phosphoryl guanidine groups reverse MDR1-mediated multiple drug resistance of tumor cells, Molecular Therapy Nucleic Acids, 27, 211-226 (2021).
[8] Zhang L, Liang XH, De Hoyos CL, Migawa M, Nichols JG, Freestone G, Tian J, Seth PP, Crooke ST. The Combination of Mesyl-Phosphoramidate Inter-Nucleotide Linkages and 2′-O-Methyl in Selected Positions in the Antisense Oligonucleotide Enhances the Performance of RNaseH1 Active PS-ASOs. Nucleic Acid Ther. 32(5):401-411 (2022). doi: 10.1089/nat.2022.0005.
[9] Anand P, Zhang Y, Patil S, Kaur K. Metabolic Stability and Targeted Delivery of Oligonucleotides: Advancing RNA Therapeutics Beyond The Liver. J Med Chem. 68(7), 6870-6896 (2025). doi: 10.1021/acs.jmedchem.4c02528. Epub 2025 Jan 8. PMID: 39772535; PMCID: PMC11998008.
[10] https://www.regulations.gov/document/FDA-2026-D-1256-0002